BMJ 2002;324:466-469 ( 23 February )
Clinical review
Regular reviewPeripheral neuropathy
Richard A C Hughes
Department of Neuroimmunology, Guy's Campus, Guy's, King's, and St Thomas's School of Medicine, London SE1 1UL
Peripheral neuropathy is common, often distressing, and sometimes disabling or even fatal. The population prevalence is about 2400 per 100 000 (2.4%), rising with age to 8000 per 100 000 (8%).1 In Europe the commonest cause is diabetes mellitus, which can produce painful neuropathy, disabling foot ulcers, and death from autonomic neuropathy. Leprosy is still prevalent in Africa, India, and South East Asia. This review explains how general practitioners can approach the first level of diagnosis and warn patients about what lies ahead after referral to a specialist.
| Summary
points
|
| | Methods |
|---|
I searched
Medline from January 1991 until September 2001 using the terms "peripheral
neuropathy" and "guideline." The search yielded 11 references,
including useful guidelines for the diagnosis and management of diabetic
peripheral neuropathy,2 but no guidelines on the
diagnosis and management of generic peripheral neuropathy. This article
offers a personal approach to the management of generalised peripheral
neuropathy from the perspective of a neurologist with a special interest
in the topic. The recommendations also take account of reviews published
by authorities in peripheral neuropathy (see educational resources)
and a recent audit of a Dutch departmental guideline that showed the
value of investigating common causes before doing electrophysiological
tests.3
| | Diagnosis |
|---|
Patients with peripheral neuropathy may present with altered sensation, pain, weakness, or autonomic symptoms. The clinical features vary widely and may resemble myelopathy, radiculopathy, muscle disease, or even hyperventilation. Identifying a neuropathy in patients with coexistent problems can therefore be difficult. The symptoms usually begin in the toes before the fingers and spread proximally.
The classic picture of advanced
polyneuropathy with distal wasting and weakness, absent tendon reflexes, and glove
and stocking sensory loss should be easy to recognise. The clinical
features allow acute symmetrical peripheral neuropathy, chronic symmetrical
peripheral neuropathy, and multiple mononeuropathy to be distinguished,
each with a different differential diagnosis.
| | Acute symmetrical peripheral neuropathy |
|---|
Acute symmetrical peripheral neuropathy is rare but important because the commonest cause is Guillain-Barré syndrome, which can be fatal. The table gives other causes. Common early symptoms are distal paraesthesiae and proximal or distal weakness occurring one to two weeks after a respiratory or gastrointestinal infection. Traditionally, the reflexes are absent, but their retention during the first hours of the illness has led many patients to be dismissed as "hysterical." Once a patient loses the ability to walk and develops facial and bulbar weakness the diagnosis becomes obvious. The rapid progression of sensory or motor deficit requires emergency investigation. Patients usually have to be admitted to hospital because of the danger of respiratory failure. Early treatment should stop the pathological process before axonal dysfunction becomes irreversible.
|
Guillain-Barré syndrome is usually due to acute inflammatory demyelinating polyradiculoneuropathy caused by an autoimmune response directed against the Schwann cells or myelin. Some cases are due to acute axonal neuropathy, in which glycolipid in the axolemma is targeted. In both forms, treatment with intravenous immunoglobulin hastens recovery and reduces the long term disability and is more convenient than plasma exchange.4 A recent trial suggests that combination treatment with steroids is more effective than intravenous immunoglobulin alone, but the full results are awaited.5
|
| | Multiple mononeuropathy |
|---|
Acute multiple mononeuropathy is also a neurological emergency because the commonest cause is vasculitis (box 1). Prompt treatment with steroids may prevent further irreversible nerve damage. If multiple mononeuropathy develops in a patient with an established connective tissue disorder (such as rheumatoid arthritis, systemic lupus erythematosus, polyarteritis nodosa, or Churg-Strauss syndrome) it is reasonable to conclude that vasculitis is the cause. Steroids are the main treatment, with cyclophosphamide being added depending on the severity and general medical condition.
Sometimes peripheral neuropathy is the presenting or sole feature of vasculitis.
In this case, vasculitis can be diagnosed only by nerve biopsy. 6 7 In addition, recent biopsy studies indicate
that diabetic amyotrophy is due to microvasculitis in the lumbosacral
plexus. It presents acutely with pain, weakness, and then wasting in
one or both quadriceps muscles.6-8
| | Chronic symmetrical peripheral neuropathy |
|---|
Most peripheral neuropathies are chronic and usually develop over several months. Diagnosis of the underlying cause may require three stages of investigation. Any history of a general medical disorder could be relevant. Patients should always be asked about alcohol consumption, toxin exposure (insecticides, solvents), and drugs. They should also have a full examination, including breasts and genitalia, to exclude underlying carcinoma.
|
The commonest causes of neuropathy can be identified from the history,
examination, and simple stage 1 investigations (box 2). Sometimes the neuropathy is predominantly sensory and subacute
with ataxia that is worse in the dark because of loss of large fibre
function and postural sensation. This pattern is produced by some drugs
(such as cisplatin), an underlying neoplasm, Sjögren's syndrome, or
idiopathic sensory neuronopathy. If other members of the family have
similar symptoms, pes cavus, or claw toes, the patient may have hereditary
motor and sensory neuropathy or Charcot-Marie-Tooth disease, which
is usually autosomal dominant. Difficulty with walking in childhood
also suggests a hereditary neuropathy. If patients have a clear cause
for their neuropathy and a typical clinical picture, treatment![]() for instance, of diabetes
mellitus or alcohol misuse
for instance, of diabetes
mellitus or alcohol misuse![]() can be started without further
investigation.
can be started without further
investigation.
Second stage investigations
If
the cause of the neuropathy is not clear from the stage 1 investigations
or is atypical, the patient should be referred to a neurologist. The
most important stage 2 investigation is neurophysiological testing
(figure). About 80% of symmetrical peripheral neuropathies are axonal
and are due to gradual dying back of the axons. In the remaining 20%
(demyelinating neuropathies) most of the damage is to the myelin, although
axonal degeneration often occurs as the disease advances. The other
second stage investigations (box 2) are simple outpatient tests for the commonest
causes of peripheral neuropathy.
|
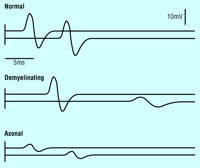
Third stage investigations
The choice of
third stage investigation will depend on whether neurophysiological
testing has shown the neuropathy to be demyelinating or axonal.
Demyelinating
neuropathy
The causes of demyelinating neuropathy are limited
(box 3). If the slowing of nerve conduction affects all
nerves roughly equally the diagnosis is likely to be the demyelinating
form of Charcot-Marie-Tooth disease (type 1). Seventy per cent of
such patients have a duplication of the gene for a 22 kDa peripheral
nerve myelin protein on chromosome 17. The duplication causes
overexpression of the protein. The clinical picture ranges from classic
pes cavus with inverted champagne bottle legs to scarcely detectable
clawing of the toes. Different mutations of the same protein and of
other myelin proteins cause a similar clinical picture. Genetic counselling
and prenatal diagnosis can be offered.
|
Chronic axonal neuropathy
Axonal polyneuropathy can be sensory or sensory and motor. It has
many causes, which will often be suggested by the history or examination.
The third stage investigations (box 4) should show the less common general medical disorders
and identify cases of diabetes mellitus that were not detected by the
fasting blood glucose test.11 Nerve biopsy should usually be done
only on patients with distressing neuropathy in whom it might lead
to useful treatment.12 In an audit of 50 cases the biopsy confirmed
the diagnosis in 70%, affected management in 60%, and caused persistent
pain in 33% of patients.12 Biopsy should be done in a specialist centre
and only when the diagnosis cannot be made in any other way. Specimens
are usually taken from the sural nerve under local anaesthetic. Vasculitis
is the diagnosis most likely to be found.
|
| | Treatment |
|---|
Any underlying
medical cause of peripheral neuropathy, such as diabetes mellitus or vitamin B-12
deficiency, should be treated. Chronic inflammatory demyelinating polyradiculoneuropathy
is important to recognise because it is treatable. Corticosteroids
are usually used initially as they are the cheapest treatment, but
the condition also responds to intravenous immunoglobulin, plasma exchange,
and some immunosuppressant drugs.9 The uncommon variant, multifocal motor
neuropathy, responds to intravenous immunoglobulin and possibly immunosuppressant
drugs but not to corticosteroids or plasma exchange.16 Unfortunately, no specific treatment
is available for chronic idiopathic axonal polyneuropathy.
| | Management |
|---|
Preventive and palliative treatments include foot care, weight reduction, and sensible shoes, boots, or ankle-foot orthoses. Patients with severe leg weakness may need sticks, crutches, or a walking frame. Physiotherapists are best placed to prescribe these aids, which may need to be adapted to take account of weakness of the hands. Simple wrist splints can help weak wrist extension. More complex splints for weak fingers and hands are usually cumbersome and rarely used. Disabled patients require help from a multidisciplinary team including an occupational therapist, who can advise on special utensils and home adaptations. Some drugs help. Sildenafil may correct erectile impotence. In the United Kingdom, the NHS will pay if the neuropathy is due to diabetes mellitus.
Patients with neuropathy may experience pain, which can be severe and out of proportion to any sensory or motor deficit. Painful neuropathy is difficult to treat. The most useful drugs are anticonvulsants, especially gabapentin and carbamazepine, and tricyclic antidepressants, especially amitriptyline. The opioid-like analgesic tramadol has also been shown to be useful in randomised controlled trials.17
| Additional
educational resources Thomas PK, Ochoa J. Clinical features and differential diagnosis. In: Dyck PJ et al, eds. Peripheral neuropathy. Philadelphia: WB Saunders, 1993:749-74. Asbury AK, Thomas PK. The clinical approach to neuropathy. In: Peripheral nerve disorders Oxford: Butterworth-Heinemann, 1995:1-28. Hughes RAC. Management of chronic peripheral neuropathy. Proc R Coll Physicians Edinb 2000;30:321-7. Sabin TD. Generalized peripheral neuropathy: symptoms, signs, and syndromes. In: Cros D, ed. Peripheral neuropathy. A practical approach to the diagnosis and management. New York: Lippincott, Williams and Wilkins, 2001: 3-20. Patient information Guillain-Barré
Syndrome Support Group (http://www.gbs.org.uk/) Peripheral Neuropathy Trust
(http://www.neuropathy-trust.org/) CMT
United Kingdom (http://www.cmt.org.uk/) |
| | Acknowledgments |
|---|
I thank David Hughes, Haider Katifi, Michael O'Brien, Mary Reilly, and Wolfgang Schady for reading the manuscript and Kerry Mills for providing the figure.
| | Footnotes |
|---|
Competing interests: RACH is coordinating editor of the Cochrane Neuromuscular Disease Review Group.
| | References |
|---|
| 1. | Martyn
CN, Hughes RAC. Epidemiology of peripheral neuropathy. J Neurol Neurosurg Psychiatry
1997; 62: 310-318 |
| 2. | Boulton AJ, Gries FA,
Jervell JA. Guidelines for the diagnosis and outpatient management of diabetic
peripheral neuropathy. Diabet Med 1998; 15: 508-514 |
| 3. | Rosenberg NR, Portegies
P, de Visser M, Vermeulen M. Diagnostic investigation of patients with chronic
polyneuropathy: evaluation of a clinical guideline. J Neurol Neurosurg Psychiatry
2001; 71: 205-209 |
| 4. | Hughes RAC, Raphael J-C, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome (Cochrane review). Cochrane Database Syst Rev 2001;(3):CD20014. |
| 5. | Van Koningsveld R, van
der Meché FGA, Schmitz PIM, van Doorn PA. Combined therapy of intravenous immunoglobulin
and methylprednisolone in patient with Guillain-Barré syndrome. J Peripheral
Nervous System 2000; 6: 186-187 |
| 6. | Davies L, Spies JM, Pollard JD, McLeod JG. Vasculitis confined
to peripheral nerves. Brain 1996; 119: 1441-1448 |
| 7. | Dyck PJ, Berstead TJ,
Conn DL, Stevens JC, Windebank AJ, Low PA. Non systemic vasculitic neuropathy.
Brain 1987; 110: 843-854 |
| 8. | Dyck PJB, Norell JE,
Dyck PJ. Microvasculitis and ischemia in diabetic lumbosacral radiculoplexus neuropathy.
Neurology 2000; 53: 2113-2121 |
| 9. | Saperstein
DS, Katz JS, Amato AA, Barohn RJ. Clinical spectrum of chronic acquired demyelinating
polyneuropathies. Muscle Nerve 2001; 24: 311-324 |
| 10. | Yan WX, Archelos JJ,
Hartung H-P, Pollard JD. P0 protein is a target antigen in chronic inflammatory
demyelinating polyradiculoneuropathy. Ann Neurol 2001; 50: 286-292 |
| 11. | Russell JW, Feldman
EL. Impaired glucose tolerance |
| 12. | Gabriel CM, Hughes
RAC, Howard R, Saldanha G, Bensa S, Kinsella N, et al. Prospective study of the
usefulness of sural nerve biopsy. J Neurol Neurosurg Psychiatry 2000; 68:
442-446 |
| 13. | Notermans NC, Wokke JHJ, Van der Graaf Y, Franssen H, Van Dijk
GW, Jennekens FGI. Chronic idiopathic axonal polyneuropathy: a five year follow
up. J Neurol Neurosurg Psychiatry 1994; 57: 1525-1527 |
| 14. | Schenone A, Mancardi
GL. Molecular basis of inherited neuropathies. Curr Opin Neurol 1999; 12:
603-616 |
| 15. | Reilly MM. Classification
of the hereditary motor and sensory neuropathies. Curr Opin Neurol 2000;
13: 561-564 |
| 16. | Nobile-Orazio E. Multifocal
motor neuropathy. J Neuroimmunol 2001; 115: 4-18 |
| 17. | Sindrup SH, Jensen
TS. Efficacy of pharmacological treatments of neuropathic pain: an update and
effect related to mechanism of drug action. Pain 1999; 83: 389-400 |
© BMJ 2002
| Home | Help | Search/Archive | Feedback | Table of Contents |